
The United States Food and Drug Administration (FDA) categorizes medical devices into three classes: Class I, Class II, or Class III. The FDA classifies medical devices based on their risk to patient safety.
Examples of FDA Class I medical devices include tongue depressors, manual stethoscopes, and bandages. FDA Class II medical devices contain wheelchairs, contact lenses, and blood glucose meters. FDA Class III medical device examples encompass pacemakers, defibrillators, and artificial hips.
The FDA recommends methods for determining the classification of medical devices: by searching their database using the device’s product code or by comparing new devices to similar, previously approved ones.
Similar to the FDA’s tiered system, the European Union Medical Device Regulation (EU MDR) categorizes devices based on risk, establishing requirements for patient safety and market approval.
Each medical device classification has distinct requirements, from general controls to rigorous premarket approvals.
Quality Management System (QMS) software streamlines the regulatory approval processes for medical devices. An increasing number of companies are adopting QMS solutions for efficient quality management and to ensure compliant processes.
SimplerQMS provides fully validated QMS software, according to GAMP 5, tailored to the needs of medical device companies. To learn more about how our QMS software streamlines quality management processes, schedule a demo with our quality solution specialist.
What Are FDA Medical Device Classes?
The FDA categorized medical devices into three classes: Class I, Class II, and Class III.
Medical device classes are a tiered categorization scheme based on potential risks to patients.
Class I medical devices present the lowest risk with minimal potential for harm. Class II medical devices are at moderate risk, having a higher risk than class I devices. Class III medical devices present the highest risk, encompassing devices that sustain or support life.
The medical device classification directs the proportionate regulatory controls for safe and effective market availability. The classification system defines progressively stricter controls as the risk level increases.
By thoroughly understanding the FDA’s medical device classification system, medical device manufacturers can streamline their development process, ensure compliance with relevant requirements, and ultimately deliver safe and effective medical devices to market.
What Is an FDA Class I Medical Device?
FDA Class I medical devices encompass the lowest-risk category of devices. Class I medical devices pose minimal potential for harm to users and typically require the least regulatory control compared to higher-class devices.
Out of all the regulated medical devices, 35% are considered to be Class I medical devices, according to the FDA Center for Devices and Radiological Health (CDRH) in 2020.
What Are FDA Class I Medical Device Examples?
Examples of medical devices in Class I are given below.
- Plasters
- Surgical mask
- Manual stethoscopes
- Electric toothbrushes
- Enema kits
- Bedpans
- Tongue depressor
- Oxygen mask
- Hospital beds

What Is an FDA Class II Medical Device?
FDA Class II medical devices comprehend devices deemed to pose a moderate risk to users.
Class II medical devices necessitate additional regulatory controls to mitigate potential risks and ensure their safety and effectiveness when compared to the minimal potential for harm associated with Class I medical devices.
Of all approved devices on the market, 53% are considered Class II medical devices, according to the FDA CDRH in 2020.
What Are FDA Class II Medical Device Examples?
Examples of medical devices in Class II are given below.
- Powered wheelchairs
- Pregnancy test kits
- Surgical gloves
- Catheters
- Blood pressure cuffs
- Blood transfusion kits
- Absorbable sutures
- Syringes
- Contact lenses

What Is an FDA Class III Medical Device?
FDA Class III medical devices encompass devices with the highest potential for harm. Class III devices sustain or support life, are implanted, or present a potentially unreasonable risk of illness or injury.
Class III medical devices face the strictest regulatory requirements compared to their lower-class counterparts due to their potential for significant harm. Class III medical devices demand rigorous premarket approval processes with extensive data on safety and efficacy, whereas Class I and Class II medical devices require general and special controls, respectively.
Of all the approved medical devices, 9% are considered Class III, according to the FDA CDRH in 2020.
What Are FDA Class III Medical Device Examples?
Examples of medical devices in Class III are given below.
- Defibrillators
- Implantable pacemakers
- Implanted prosthetics
- Breast implants
- Cochlear implants
- High-frequency ventilators
- Fetal blood sampling monitors

How To Classify Your Medical Device According to FDA Classification Rules?
There are different methods by which medical device manufacturers can determine the class of their medical devices in the US market.
Key methods endorsed by the FDA for effectively classifying medical devices are listed below.
- Device Classification Panel
- Product Classification Database
- Similar Devices by Clearance or Approval
- Similar Devices by Device Listing
Method 1: Device Classification Panel
The FDA organizes medical device classifications into 16 specialized panels covering diverse medical specialties, such as cardiovascular, immunology, orthopedic, ear, nose, and throat, among others.
The FDA device classification panel encompasses over 1,700 distinct types of medical devices, offering a structured framework for manufacturers to identify applicable classifications and requirements.
Medical devices can be classified by finding the corresponding description of the device in Title 21 of the Code of Federal Regulations (CFR), Parts 862-892.
Manufacturers who know the device panel to which their device belongs can go directly to the listing for that panel and identify the device and the corresponding regulation.

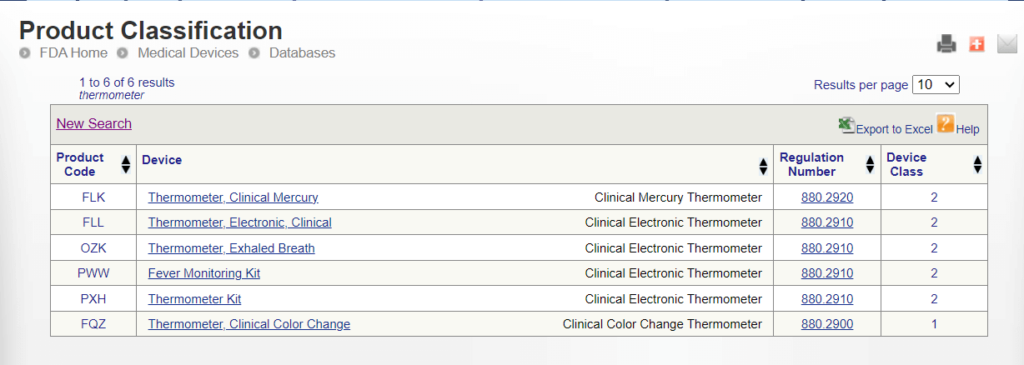
Method 2: Product Classification Database
Medical device manufacturers can search directly for an appropriate product classification in the FDA’s public Product Classification Database.
Manufacturers can search the database using either the specific device product code or a portion of the device’s name.
FDA product codes are three letters used to identify specific medical devices within the FDA’s regulatory framework. Manufacturers who know their device’s product codes can find the device class and applicable requirements directly.
Method 3: Similar Devices by Clearance or Approval
Similar devices already being marketed can be used as a model to determine the classification of new ones.
Medical device manufacturers can work backward by determining how a similar device has been classified based on its clearance or approval to market. Manufacturers can then decide if the same classification applies to the new proposed device.
For this method, manufacturers can search the different databases depending on the device’s marketing authorization route.
- 510(k) Clearance: The 510(k) is a premarket submission made to the FDA to demonstrate that the device to be marketed is safe and effective. The Premarket Notification 510(k) database encompasses most Class II devices requiring 510(k) clearance from the FDA before being legally marketed.
- Premarket Approval (PMA): The PMA is the FDA process of scientific and regulatory review to evaluate the safety and effectiveness of Class III medical devices. Manufacturers can find the approved Class III medical devices in the Premarket Approval database.
- De Novo: The De Novo request provides a marketing pathway to classify novel medical devices of low to moderate risk. Innovative Class I and II devices can be found in the De Novo database.
Method 4: Similar Devices by Device Listing
Medical device manufacturers can access a comprehensive listing of all registered medical devices and search for similar devices to be used as a classification model.
Most Class I and some Class II medical devices may not be listed in the clearance or approval databases because they are exempt and do not require the FDA’s review before marketing.
However, device registration and listing are general controls mandated for all medical devices in the US. Thus, the Establishment Registration and Device Listing database provides the list and classification of all currently marketed devices.
What Is the Difference Between FDA and EU MDR Medical Device Classification?
The FDA takes a broader approach to classifying medical devices than the EU MDR.
Both the FDA and EU MDR share a focus on risk-based classification. However, the MDR system employs more classes, offering a more stringent and detailed approach to classifying medical devices.
According to the EU MDR medical device classification, medical devices are categorized into Class I, IIa, IIb, and III. Moreover, MDR subdivides Class I devices in Is, Im, and Ir based on specific sterility, measurability, or reusability characteristics, respectively.
What Are the Different FDA Medical Device Approval Pathways?
The approval pathway for a medical device in the US is directly linked to its classification by the FDA.
NOTE
This article will briefly discuss the medical device regulatory pathways according to FDA. Please always refer to the official FDA regulation applicable to your medical device for comprehensive information.
The regulatory control level directly correlates with a medical device’s designated risk class. Class I medical devices, representing the lowest potential risk to patients, are subject to the most minimal regulatory controls, such as Good Manufacturing Practices (GMP) and labeling requirements.
On the other hand, Class III medical devices, posing the highest potential risk, require the most stringent regulatory requirements, including premarket approvals and extensive post-market surveillance.
The regulatory controls for each device class include:
- Class I: General Controls and Premarket Notification 510(k)
- Class II: General Controls, Special Controls, and Premarket Notification 510(k)
- Class III: General Controls, Special Controls, and Premarket Approval (PMA)
Below is a visual overview of the medical device FDA approval pathway.

FDA Class I Medical Device Approval Pathway
The pathway to market for Class I medical devices encompasses General Controls and, in some cases, Premarket Notification 510(k).
General controls are basic requirements that every medical device manufacturer must comply with, regardless of class. General controls cover aspects such as good manufacturing practices, quality management systems, labeling, and adverse event reporting.
Many Class I medical devices are exempt from Premarket Notification 501(k) due to their low risk and well-established technology. However, some Class I devices, particularly those that are novel or raise potential safety concerns, may require a 510(k) to demonstrate their substantial equivalence to a similar device already cleared by the FDA.
FDA Class II Medical Device Approval Pathway
The pathway for Class II medical devices within the FDA regulatory framework faces General Controls, Special Controls, and Premarket Notification 510(k).
Class II medical devices must comply with General Controls, similar to Class I devices, and may or may not need to submit a Premarket Notification 510(k) based on their potential risk to user safety. However, due to Class II devices’ moderately higher potential risk, additional Special Controls must be in place.
Special Controls are regulatory requirements for Class II devices for which general controls alone are insufficient to provide reasonable assurance of the safety and effectiveness of the device.
Special controls are usually device-specific and include:
- Performance standards
- Post-market surveillance
- Patient registries
- Special labeling requirements
- Premarket data requirements
- Guidelines
FDA Class III Medical Device Approval Pathway
The regulatory pathway for Class III medical devices encounters the most stringent requirements, including General Controls, Special Controls, and Premarket Approval (PMA).
Class III devices are subject to the foundational frameworks of General Controls, just like Class I and II devices, ensuring adherence to good manufacturing practices. If necessary, Class III medical devices must also comply with Special Controls.
The defining requirement for Class III devices is the Premarket Approval (PMA). The PMA approval is based on a determination by the FDA that the device documentation contains sufficient valid scientific evidence to assure that the device is safe and effective for its intended use.
Data requirements for PMA include the following.
- Non-clinical Laboratory Studies Section: The non-clinical laboratory studies section comprises data and analyses from microbiology, toxicology, immunology, biocompatibility, stress, wear, and shelf-life studies, along with other relevant testing. The non-clinical laboratory studies section must thoroughly assess the device’s safety and efficacy profile.
- Clinical Investigations Section: The clinical investigations section comprehends the data from the clinical investigations, encompassing study protocols, safety and efficacy outcomes, adverse events, device failures and replacements, patient information and complaints, statistical analyses, and any other pertinent information gathered from the research.
What Is the Role of QMS Software in Facilitating FDA Medical Device Approval?
The Quality Management System (QMS) software facilitates FDA approval by streamlining documentation, risk management, and compliance management processes throughout medical devices’ entire development and approval lifecycle.
QMS software organizes essential documents like design input and output, design verification and validation, design review, procedures, and records, ensuring easy access and audit readiness. Pre-defined workflows and notifications help manage risks and audits, showcasing continuous improvement efforts to the FDA.
The medical device QMS software supports manufacturers in achieving compliance with regulatory requirements, including 21 CFR Part 11 and 820, GMP, ISO 13485:2016, MDR, and IVDR, among others.
SimplerQMS provides QMS software fully validated according to GAMP 5. Our software solution ensures compliance with the software validation requirements of FDA 21 CFR Part 820.70.
The SimplerQMS solution supports companies in achieving compliance with regulatory requirements by providing comprehensive QMS process support. SimplerQMS supports all QMS processes, such as document control, change management, training, supplier management, audit management, nonconformances, and more. The system offers document collection and archive capabilities to structure data and facilitates regulatory submissions.
Facilitate your journey to FDA approval with SimplerQMS. Book a personalized demo with one of our Quality Solution Consultants today and explore how the SimplerQMS solution can optimize your quality management processes and get to market faster.