
The Medical Device Single Audit Program (MDSAP) is a program that allows the conduct of a single regulatory audit of a medical device manufacturer that satisfies the relevant requirements of multiple regulatory authorities.
Audits are conducted by Auditing Organizations approved by the Regulatory Authorities of the participating MDSAP members, including Australia, Brazil, Canada, Japan, and the United States.
The MDSAP is a way for medical device manufacturers to target multiple new markets and minimize business disruptions that can happen with several different regulatory audits.
In this article, we will discuss the MDSAP purpose, benefits, the participating regulatory authorities, the typical audit process, and how a modern Electronic Quality Management System (eQMS) can help your company prepare for the MDSAP.
An eQMS is one of the best ways to prepare for the MDSAP. SimplerQMS provides eQMS software for medical device companies to manage quality management processes, reduce workflow disruption, and make compliance easier to achieve. To learn more about how SimplerQMS can help your company book a demo and talk to one of our Quality Solution experts.
We will cover the following topics:
- What Is Medical Device Single Audit Program (MDSAP)?
- What Is the Purpose of MDSAP?
- What Are the Benefits of the MDSAP?
- Which Regulatory Authorities Are Participating in MDSAP?
- MDSAP Audit Cycle
- MDSAP Audit Process
- MDSAP Key Considerations
- Frequently Asked Questions
- The Role of eQMS Software in Preparation for MDSAP
What Is Medical Device Single Audit Program (MDSAP)?
The MDSAP is a program that allows medical device companies to face a single third-party audit conducted by authorized Auditing Organizations to satisfy the regulatory requirements of its participating members.
With MDSAP, medical device companies undergo a single audit for compliance with the regulatory requirements of five medical device markets: Australia, Brazil, Canada, Japan, and the United States.
What Is the Purpose of MDSAP?
The purpose of MDSAP is to accelerate international medical device regulatory harmonization and to promote an efficient and effective regulatory model.
The program was developed to enable a regulatory overview of medical device manufacturers’ QMS while reducing the regulatory burden on the industry. It aims to promote worldwide alignment of international requirements.
For instance, a medical device company sells 3-dimensional microscopes for reconstructive surgery. Complying with the requirements of MDSAP allows the company to market and sell the device in five major international markets.
What Are the Benefits of the MDSAP?
Being MDSAP certified presents many benefits to medical device manufacturers, patients, and regulatory authorities.
Benefits for medical device manufacturers:
- Single comprehensive audit: medical device companies can undergo a single audit process, not several individual audits. Being MDSAP certified reduces business disruptions and saves resources, such as time, finances, and training.
- Commitment to quality: MDSAP certification emphasizes to regulatory authorities and customers the company’s commitment to quality and compliance with international requirements.
- Predicted audit schedules: MDSAP has a comprehensive schedule for a three-year period. So, medical device companies have a clear understanding of their timeframes and resources.
Benefits for patients and end-users:
- Patient safety: By complying with MDSAP documents, medical device companies assure patients and end-users that all devices are safe and effective for the intended use.
Benefits for regulatory authorities:
- Multiple compliances: Regulatory authorities participating in the MDSAP are assured that auditing has been conducted in accordance with the agreed requirements. This reduces the workload of individual regulatory authorities.
Which Regulatory Authorities Are Participating in MDSAP?
The MDSAP is a program under the International Medical Device Regulators Forum (IMDRF).
IMDRF is a voluntary group of medical device regulators from around the world and aims to accelerate international medical device regulatory harmonization and convergence.
There are several countries in the IMDRF group. However, the international partners that are participating in the MDSAP include:
- Therapeutic Goods Administration of Australia
- Brazilian Health Regulatory Agency (Anvisa)
- Health Canada
- Ministry of Health, Labour and Welfare of Japan, and Japanese Pharmaceuticals and Medical Devices Agency
- US Food and Drug Administration
Therapeutic Goods Administration of Australia
The Australian Therapeutic Goods Administration (TGA) uses MDSAP audit reports and certificates as part of the evidence for compliance with medical device conformity assessment procedures and market authorization requirements.
Medical device manufacturers audited under MDSAP may, under some circumstances, avoid routine TGA inspections.
Brazilian Health Regulatory Agency (Anvisa)
Brazilian Health Regulatory Agency (Anvisa) utilizes MDSAP reports to constitute an important input on pre-market and post-market assessment procedures.
ANVISA may use MDSAP audits in place of a pre-market inspection for manufacturers intending to put higher-risk devices on the Brazilian market. For subsequent assessments, ANVISA continues to use its regular audits and inspections.
Health Canada
Health Canada started accepting MDSAP certificates at the beginning of January 2019.
This means that medical device companies do not have to undergo any additional audits in Canada regarding the QMS.
Japanese Regulatory Authorities
The Japanese Regulatory Authorities (Ministry of Health, Labour and Welfare of Japan, and Japanese Pharmaceuticals and Medical Devices Agency) accept MDSAP audit reports exempting some manufacturing sites from on-site inspection.
This reduces medical device manufacturers’ burden in QMS audit processes and may lead to the reduction of documentation required to be submitted for off-site inspection as well.
US Food and Drug Administration
The US FDA will accept the MDSAP audit reports as a substitute for FDA Establishment Inspection Reports (EIR).
The EIR describes what was observed during the FDA investigator’s visit to the company site, from introductions to inspectional observations.
It is worth mentioning that when US requirements are audited, FDA 21 CFR Part 820 for QMS regulation should appear on the certification document.
MDSAP Audit Cycle
The complete MDSAP has a three-year audit cycle consisting of the following:
- An Initial Audit (the Initial Certification Audit)
- Partial Surveillance Audits in years 2, 3
- Complete Re-Audit in year 3

The MDSAP audit model will review medical device companies’ QMS compliance with ISO 13485:2016 as well as the individual regulatory requirements of the participating countries where companies wish to market finished devices.
The Initial Audit will comprehensively audit the Medical Device Quality Management System.
It consists of two audit stages. The purpose of the Stage 1 Audit is a complete review of the QMS documentation required by ISO 13485:2016 and to evaluate preparedness for the next stage audit.
The goal of the Stage 2 Audit is to evaluate QMS implementation and effectiveness in accordance with ISO 13485:2016 and the requirements of the participating regulatory authorities.
A series of Surveillance Audits aims to ensure that all applicable requirements mentioned in the Initial Audit are continuously inspected during the three-year cycle.
Finally, a Re-audit (Recertification audit) aims to confirm the continued relevance, applicability, and suitability of the medical device company’s QMS as a whole. It can include a Stage 1 Audit if your QMS has undergone a critical change that was not sufficiently assessed previously.
In addition to these audits, medical device companies can undergo Special Audits that are not part of the planned audit cycle. A Special Audit may be conducted in response to an application to extend the scope of an existing certification, for example.
Another important audit is an Unannounced Audit. This audit can be conducted anytime during the three-year audit cycle. During this audit, the Auditing Organizations will randomly select a representative product and check whether it has been manufactured in accordance with your company’s QMS.
Recommended Reading
MDSAP Audit Process
This article will briefly discuss the MDSAP audit process.
We will also give examples of how an Electronic Quality Management System (eQMS), like SimplerQMS, can help your company successfully prepare for the MDSAP certification.
NOTE
This article does not explain all the details related to MDSAP. Do follow the official standards and requirements that apply to your company. The complete pieces are provided on official websites, such as the FDA – MDSAP Audit Procedures and Forms.
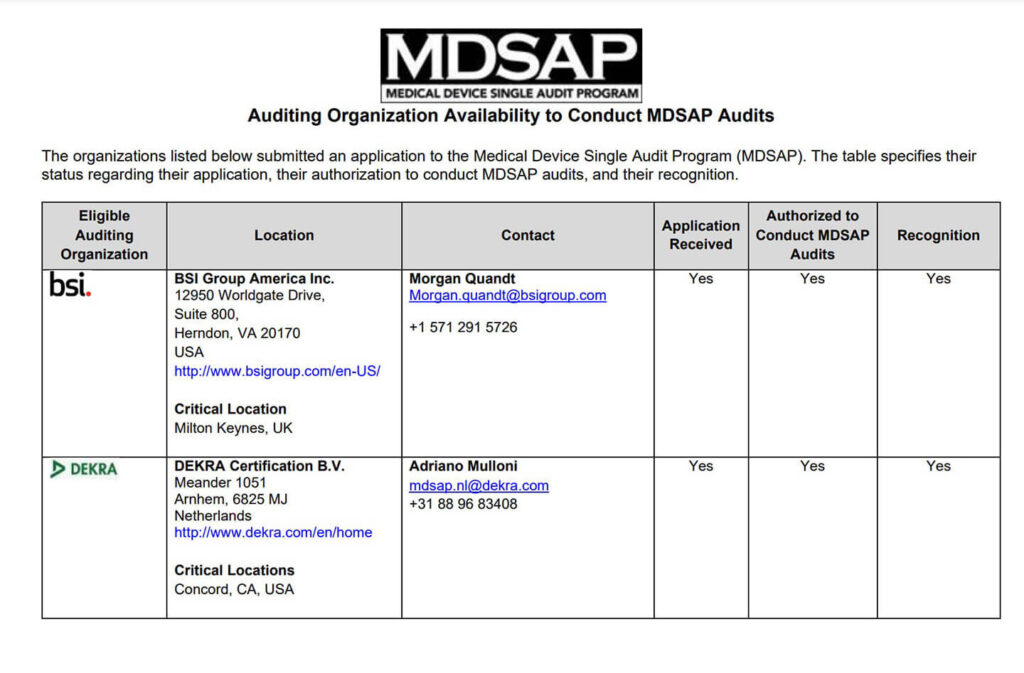
As stated before, the International Medical Device Regulators Forum (IMDRF) has recognized third-party auditors to conduct the MDSAP audit for medical device companies, known as Auditing Organizations (AO).
Each participating member has a list of approved AOs. For instance, the FDA has a list of recognized auditing organizations. Check each regulatory authority website to find the authorized AO list of your interest.
Four primary processes, one enabling process, and two supporting processes are part of the MDSAP audit.
Primary processes:
- Management
- Measurement, Analysis, and Improvement
- Design and Development
- Production and Service Controls
Enabling process:
- Purchasing
Supporting processes:
- Device Marketing Authorization and Facility Registration
- Medical Device Adverse Events and Advisory Notices Reporting
The auditors will start with the Management Process and then proceed to the second primary process, and so on.
The two supporting processes will fulfill the regulatory requirements of the participating jurisdictions.
Management Process
The top management of medical device companies must ensure that:
- Sufficient resources are provided for device design and manufacturing
- The quality management system is proper and effective
- Monitor the QMS and make adjustments when needed
- Define, document, implement, and maintain the QMS requirements via a representative.
It is essential that top management show commitment to quality. The quality commitment must be communicated across the company to assure employees and third-party stakeholders, such as the regulatory authorities and end-users, that finished medical devices are safe and efficient for their intended purpose.
The quality data obtained from different sources within and outside the company must be analyzed. With this information, companies can measure preparedness for the audit, make changes in processes if required, provide additional resources, and so on.
Collating data into a single source of truth is much easier using an eQMS system paired with powerful document controls such as SimplerQMS. It allows medical device companies to gather data from all departments and processes in a single cloud-based system.
Documents and related information can be easily linked, versioned, routed for review/approval, and retrieved during the audits.
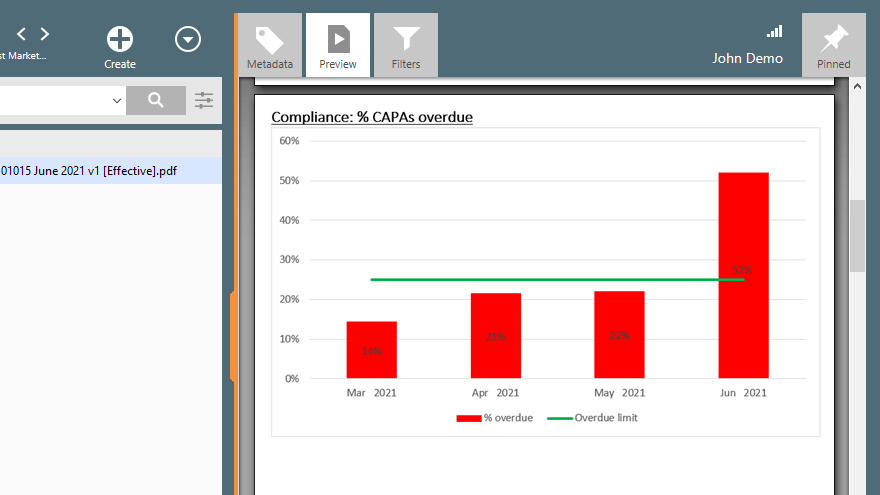
Measurement, Analysis, and Improvement Process
In MDSAP, the QMS emphasizes identifying existing and potential causes of product and quality issues.
To take appropriate and effective corrective and preventive actions, these causes must be identified.
All these processes are carried out under the Measurement, Analysis, and Improvement process. Also, outcomes must be documented and reported to the relevant regulatory authority.
An eQMS solution helps medical device manufacturers manage nonconformance, complaint, and CAPA processes.
SimplerQMS software solution allows companies to keep track of all nonconformances, complaint resolution progress, CAPA status, and so on to easily manage and document all data with just a few clicks.
Design and Development Process
The Design and Development Process aims to control the design of a medical device and to assure that the device meets user needs, intended use, and its specified requirements.
The key steps that medical device companies are expected to follow are:
- Design and development planning: Have updated and approved plans for all manufactured medical devices.
- Design input: Have design input procedures to ensure the initial design for a given medical device corroborates with its actual use. A design must be reevaluated if there is ambiguity or incompleteness.
- Design output: Ensure the design output of every medical device conforms to the original design input.
- Design review: Monitor and conduct formal documented reviews of specific devices at selected intervals. Document the results in the Design History File (DHF).
- Design verification: Ensure the conformance of design output with design input with established QMS protocols.
- Design validation: Have compatibility between devices and related software with the intended use. Companies can use either stimulated or actual use conditions to validate.
- Design transfer: Ensure the device design is translated into correct product specifications.
- Design changes: Control design changes to improve the design of a medical device.
- Design history file: Use the medical device’s DHF to prove the device was manufactured following approved plans.
The design and development process requires creating, maintaining, validating, and circulating multiple files and documents between departments. This is easier to perform and track with an eQMS solution.
SimplerQMS’s design control software module helps medical device companies to keep all design control-related documents in one place, reducing the chances of errors due to manual paperwork.
Easily create documents using a template package based on Life Science requirements, link files to different design control archives without creating duplicates, assign tasks to specific people, drag and drop external documents inside the system, and migrate all existing files to a single cloud-based system.
Production and Servicing Controls
The purpose of auditing the Production and Servicing Control is to verify the development of processes used to manufacture products that meet specifications.
Medical device companies need to consider the processes regarding:
- Environmental controls: Any environmental changes can affect the finished device’s overall quality.
- Contamination controls: The manufacturing area of the plant and the equipment used must be clean and sterile, if applicable.
- Personnel controls: Staff health, cleanliness, and clothing need to be checked regularly.
- Building controls: Sufficient space should be available for every activity in the product lifecycle.
- Manufacturing materials control: Materials used for manufacturing specific devices must be suitably discarded if compromised in any way.
- Servicing controls: Determine if servicing activities are performed and documented following instructions and procedures.
This process emphasizes various crucial controls that are needed during the lifecycle of a medical device. Once more, an eQMS solution can help in ensuring these controls are in place.
With SimplerQMS, medical device companies can create document collections to compile technical documentation, DHF, DMR, and DHR. Our solution also allows companies to schedule periodic reviews of these documents, automatically notify relevant personnel when changes are made, and securely store all the needed documentation for each device in a cloud-based system.
Purchasing Process
The medical device company is expected to establish and maintain documented controls for planning and performing purchasing activities. This refers to third-party suppliers, including contractors and consultants, who supply components, materials, or services.
Using an eQMS solution with supplier quality management features, like SimplerQMS, facilitates selecting, evaluating, qualifying, and managing your suppliers.
You can create and maintain Approved Suppliers Lists (ASLs) and use templates based on Life Science requirements for various documents, such as contracts, surveys, evaluations, certificates, and incoming inspections. Easily assign and track supplier-related tasks while keeping the records of all suppliers secure in one place.
Recommended Reading: Medical Device Supplier Management Process (8 Steps)
Device Marketing Authorization and Facility Registration Process
Auditing these processes aim to verify that medical device companies performed the appropriate activities regarding device marketing authorization and facility registration with regulatory authorities participating in the MDSAP.
Medical Device Adverse Events and Advisory Notices Reporting
This process audit verifies that a post-market surveillance system is established and implemented in the medical device company and integrated into the QMS. Procedures and workflows are established to ensure the correct and prompt identification of adverse events.
For instance, a batch of implantable pacemakers is malfunctioning, and several healthcare facilities filed complaints. The company needs to identify the root cause of the issue, ensure it does not repeat it, and report to the relevant regulatory authority.
For that, medical device companies need the capability to continuously collect and analyze data generated from every medical device manufactured and marketed.
An eQMS, such as SimplerQMS, with in-built complaint management capabilities, can make the process easier.
For example, using SimplerQMS, companies can report incidents to regulatory authorities in case the nonconformance presents a potential risk to patients or results in injury or death.
MDSAP Key Considerations
Medical device companies planning on undergoing the MDSAP should note the following points:
- Audit duration: The audit cycle of MDSAP has three years. It consists of an Initial Audit (in two separate stages), two Surveillance audits in the next two years, and a Recertification Audit in the third year.
- Audit requirements: Since MDSAP is a comprehensive audit program involving multiple inspectors, companies should be well-prepared with trained personnel. They should be available to answer the inspectors’ queries and provide the required documents and records.
- Resources: MDSAP audits have a cost for medical device companies to be certified. Companies must have the appropriate resources.
- Documentation and record keeping: There is a need to have a robust QMS with efficient documentation and record-keeping implemented.
Traditional paper-based or hybrid QMS can present challenges, such as lost documents, delays in document management activities, lack of traceability, etc.
So, it is important to consider transitioning to an eQMS. SimplerQMS solution facilitates efficient document management. You can store all records securely in one place, easily search and retrieve documents, assign tasks, automatically notify personnel, and so on.
Frequently Asked Questions
What is the Difference Between ISO 13485:2016 and MDSAP?
The difference between ISO 13485:2016 and MDSAP is that ISO 13485:2016 is an international quality standard, and MDSAP is an audit program.
Furthermore, ISO 13485:2016 is a regulatory medical device QMS standard utilized worldwide. On the other hand, MDSAP is currently conducted in five countries: Australia, Brazil, Canada, Japan, and the United States.
Does FDA Recognize MDSAP?
The US Food and Drug Administration (FDA) is one of the participating members of the MDSAP. It accepts MDSAP audit reports as a substitute for FDA Establishment Inspection Reports (EIR) for on-site inspections.
Is the EU Part of MDSAP?
The European Union (EU) is an official observer of MDSAP and may attend MDSAP meetings, assessments, and other activities. However, MDSAP is not accepted in the EU to replace or supplement its regulatory scheme.
Who Can Perform MDSAP Audits?
MDSAP audits are performed by Auditing Organizations (AOs). These organizations are authorized by the participating Regulatory Authorities to audit under MDSAP requirements. Medical device manufacturers that wish to be MDSAP certified must refer to their national AO list.
Is MDSAP a Certification?
The MDSAP provides a certification that allows a medical device manufacturer’s QMS to be audited using a single audit and accepted in five countries: Australia, Brazil, Canada, Japan, and the United States.
What are the Principles of MDSAP?
MDSAP is based on ISO 13485:2016 requirements. In addition, medical device manufacturers must comply with specific country regulatory requirements for Australia, Brazil, Canada, Japan, and the United States, depending on the intended market.
What Processes Include the MDSAP Audit Scope?
The MDSAP audit scope has a total of seven processes.
There are four primary processes: Management Process, Measurement, Analysis, and Improvement; Design and Development; and Production and Service Controls.
One enabling process: Purchasing.
And there are two additional supporting processes: Medical Device Adverse Events and Advisory Notices Reporting and Device Marketing Authorization and Facility Registration.
The Role of eQMS Software in Preparation for MDSAP
The Medical Device Single Audit Program is a unique opportunity for medical device manufacturers to access up to five international markets with a single QMS audit.
However, medical device companies do need to extensively prepare for a comprehensive, time-consuming, and expensive audit keeping in mind the requirements of multiple jurisdictions.
When using paper-based or hybrid QMS systems, challenges may arise in the form of lost documents and records, physical storage issues, unauthorized access to files, human errors, and so on.
Considering that an MDSAP audit requires extremely meticulous documentation and record-keeping, a purpose-built medical device eQMS solution can be an excellent asset for any medical device company.
An eQMS provides traceability of documents, time-stamped audit trails, automated notifications, liking documents, assigning tasks, and so on.
Best QMS software solutions for life sciences support a vast array of core QMS processes, such as document control, change management, training management, complaint handling, supplier management, and CAPA management, that are closely related to processes evaluated during MDSAP audits.
So, investing in a robust and powerful medical device eQMS, such as SimplerQMS, can make the challenging path of preparing for an MDSAP audit much smoother and simpler.
If you are interested in calculating the investment of a QMS software solution, download our eQMS Business Case Template. It will help you understand the cost of such a solution and provide the tools to present it to senior stakeholders and decision-makers.

Final Thoughts
The medical device industry is highly regulated. Companies must comply with multiple regulatory requirements to legally market and sell their devices in different countries.
To be certified according to these requirements, medical device companies must undergo third-party audits, which can be time-consuming and expensive.
Aiming to conduct a single regulatory audit of a medical device manufacturer’s QMS that satisfies the requirements of multiple regulatory jurisdictions, the Medical Device Single Audit Program was created. It has five participating members: Australia, Brazil, Canada, Japan, and the United States.
The MDSAP is a way that medical device companies can be audited once for compliance with the standard and regulatory requirements of these five different markets.
During audits, having a paper-based or hybrid QMS can present challenges, such as lost documents, human errors, physical storage limitations, unauthorized access to documents, and so on.
An eQMS offers a solution to all those issues. And SimplerQMS Software helps medical device manufacturers to streamline all audit-related activities. You can create documents using a template package based on Medical Device requirements, link relevant documents, directly escalate nonconformances to CAPA, set up automatic reminders for tasks’ due dates, and more.
Book a personalized demo and talk to our experts if you want to learn more about how SimplerQMS can support your company QMS.
